
Our unique stories
One genetic syndrome
>19 distinct mutations to the GNB1 gene
Many unique symptoms
Incredibly infectious smiles
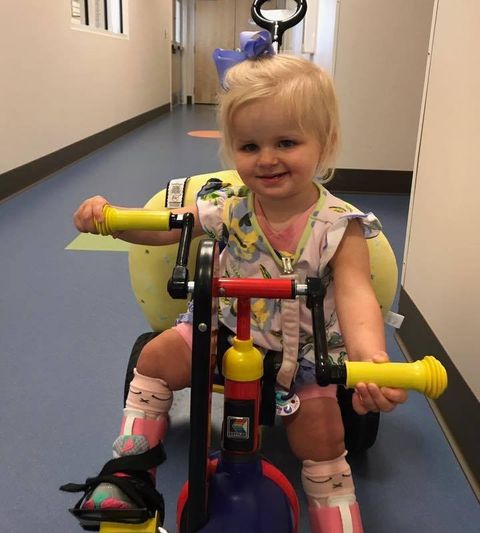
Layla
2.5 years, USA
A short back story on Layla ... from day one, she was the easiest, calmest, most quiet baby you have ever seen. My husband and I were THRILLED to have such an easy baby, because her brother who is 18 months older wasn't always so easy (to say the least)! She slept 12 hours at night from the first day we got home from the hospital. She NEVER cried. You could literally lay her down and leave her there all day and she would not make a peep. We questioned daily why she was so easy. Was something wrong? Were we not interacting with her enough because we were too busy chasing an 18 month around?
Finally, at her 3-month appointment we mentioned it to her pediatrician. He told us, he had already made note of similar concerns and was just hoping she would catch up. She wasn't doing the things a 3-month-old should be doing. She wasn't doing some things that even a newborn could do. So, at this appointment he encouraged us to work really hard with her and we would re-evaluate at the next appointment. At her next appointment, we could tell she was getting a little stronger, but she still couldn't hold her head up and she couldn't put weight on her legs, so Dr. Lee encouraged us to possibly think about going to see a neurologist. He also told us it could take months to get an appointment, so we all agreed we would go forward with this ASAP. After a very long year, appointments with a neurologist, geneticist, ophthalmologist and nephrologist and many tests we finally got our answer. GNB1. Layla had just turned a year old when we got this answer and although it seemed like forever to us, we were so thankful we got an answer at such an early age as many of these children went YEARS without this diagnosis.
So where do we go from here? I did LOTS of research and came up short. There was NO information out there regarding GNB1 except one article that referenced 13 children with this genetic mutation. I was determined to get in touch with these families or ANYONE that might be familiar with this, so I created a Facebook group and over a year I had several families join this group as they were in the same boat as us. They searched and searched for other families and came across my Facebook group that I had created and finally we were able to connect and cope together through this process.
Now at 2.5 years old, Layla is still unable to speak or talk but she continues to be the most laid back and happiest baby I have come across. Seizures are a big issue with most of these children, however, Layla has yet to have any seizures at this point. We know we have a long road ahead of us, but we are very thankful for this new foundation and the hope of what is to come for our special children!

Alice
Vivacious, happy, coffee lover, 23 years, USA
I (Denise) am the mother of a lovely, vivacious 23 year-old woman with autism. She is good natured and is noticeably happy for the most part, but has minimal communication ability, intractable seizures and motor problems.
Her autism became apparent at around two years old in 1997; she had pretty normal development to that point, but started slipping away. With her regression, she lost all language and much of the fine motor skills she had acquired. She received a diagnosis of autism at 27 months. Some notable childhood traits included a large forehead, abnormal bone development, joint hypermobility, late development of gross motor skills, an abnormal EEG, and sensory challenges (she hated the sound of a large outdoor fountain that was near our home). As a baby, she did not seem bothered by her motor challenges; she was happy and content to walk at a 13 months holding onto our hands, but did not walk alone until 17 months. Unfortunately, by age two it was apparent that her skills had stealthily declined and she experienced the disturbing loss of eye contact and initiated stereotypic behaviors like spinning and odd hand movements often seen in autism.
Like most people diagnosed with autism she was tested for chromosomal abnormalities as a child — this test was normal. A test for Rett Syndrome also came back normal.
Fourteen years later, in 2011, our family was able to get an appointment with a clinical geneticist interested in autism. He mentioned genetic testing as a future plan. Then it took until 2014 to arrive at being considered for exome sequencing. This is a genetic test that looks for mutations in that 1% protein-coding portion of the genome. Insurance gave clearance after a few months. We did the testing as a "trio," meaning that my husband, myself, and daughter all submitted blood samples. We did the testing through Stanford, where I work.
After a few months the results came back... with nothing important to tell us. There was a missense mutation found, but it was not known to be pathogenic, even I had that same mutation.
Then more than a year later, in Dec 2015, our family got an interesting phone call from the geneticist. A de novo germline mutation was found in our daughter's genome. The mutation was on chromosome 1, and concerns a gene called GNB1.
In 2015, a team at Columbia University identified the first few cases of individuals with a mutation in GNB1. Columbia then queried genetic repositories worldwide and found a total of 13 individual with this mutation. The Columbia researchers published a paper sharing their amazing findings in April 2016.
During our fateful call in 2015, our geneticist also ventured to add that he thought the GNB1 mutation was the cause of our daughter’s autism. Wow, autism explained – in our case, the explanation is an identified rare disease that has autism as one of its features.
I’ve always been driven, but now I have a mission with a solid direction. I now have knowledge of what is causing my daughter's many challenges and symptoms. Rather than addressing that amorphous thing we call "autism," I can instead direct my attention to her particular disorder and its biological pathways. Rather than treating autism, we can support research on pharmaceutical treatments that target blocking or unblocking receptors and signals that aren't working in our daughter's specific syndrome.
"Rather than addressing that amorphous thing we call 'autism,' I can instead direct my attention to her particular disorder and its biological pathways."
This mutation is noted to be disease causing (though it does not cause autism symptoms in all cases) and we know the mutation affects many bodily systems. Through the years, she has had many symptoms of some sort of unknown underlying illness – a general malaise that could not be pinpointed. We now know that her chronic hematological abnormalities through the years likely stem from the mutation and unfortunately, is an ominous symptom to follow as she ages. We can explore what her mutation does to the body’s chemistry that may influence her seizures. Her treating physicians can help us to use medicine to prevent potential health problems that are unique to people who have this mutation. She fits a particular mold, which we now recognize and better understand.
My short-term hope is to find the cause of seizures that many of the individual with the mutation experience. These seizures are often life-threatening and must be avoided. In the long term, I hope research unveils the cell signaling that is dictated by the GNB1 gene which will advance our understanding of the pathogenesis of the mutation and human biology in general.
Like everything in autism, nothing about this mutation is straightforward. Individuals with glitches in GNB1 present with a spectrum of features, from verbal to non-verbal, ambulatory to non-ambulatory, and having sensory challenges of differing degrees depending on where the mutation is located on the gene. Knowing that defects within one single gene can seed such a dizzying array of phenotypes should give us pause when we even think of “autism” as a unitary disorder.
But we also have a great deal in common, and connecting with other GNB1 families has been nothing short of amazing. I get emotional just writing about it. I see photos of the young children with this mutation, and I worry. Many have not yet developed seizures but unfortunately may do so as they age. We must assist these parents so that they can take preventive measures to avoid their child’s exposure to seizures. I feel empowered with this newfound information and motivated to nth degree to understand more.
Now, I am not a Facebook fan (in fact, I am a bit of a hater) but I must say that for our families, it’s been the best thing EVER. A parent set up a GNB1 disorder private group that now includes families from more than 10 countries. We share all sort of information about our kids that help us evaluate trajectory and interventions. We are also working on a GNB1 website that is about to go live (gnb1.org)
I want to encourage all autism families to undergo genetic testing. We must remember that as parents, we help guide our loved one’s care – so please ask for testing and follow-up on the process. Ask who you should call to follow-up on the authorization process and call monthly (I would like to encourage follow-up calls – the insurance process can be difficult and clinicians have large caseloads). Do not feel shy about asking for testing, and insisting — you are your loved one's absolute best advocate.
Scarlett
Always smiling, super-human happiness, 2 years, Australia
All our dreams came true when Scarlett Rachel Elaine was born. She was healthy, with an Apgar score of 9, and although born with a cleft palate and club foot, life seemed complete. Of course, we put these two birth defects down to coincidence and were just the happiest parents in the world.
It wasn’t until we were at a pediatrician appointment for Scarlett’s reflux, that we heard a word that would soon turn out to completely change our lives forever. Scarlett was a “HYPOTONIC” baby.
What did that mean? What is Hypotonic? Does Scarlett have Hypotonia? Scarlett was a very floppy, quiet baby, who rarely cried. Looking back, I wonder whether we were just too positive to notice, or reassured by everyone who said “every baby develops at their own pace” but really, we didn’t know any different, and we NEVER wanted to believe anything was wrong with our little girl. We pushed to go back to the pediatrician when Scarlett was 4 months old, and we realized she couldn't hold up her head, maintain eye contact, fix & follow, grab or even seeming interested in toys; and wasn't meeting all the usual milestones. It was very hard to admit that, although perfect to us in every single way, Scarlett was different and we had to help her. Over two years later and unfortunately Scarlett still can't do most of those things, but she has definitely shown a lot of progress and promise towards them.
Scarlett’s life has been a constant flow of medical appointments, tests and therapies, but also the most blissful joy and happiness. Scarlett had her first surgery at just 8 weeks of age, and has been put under general anesthetic several times. Scarlett has a number of secondary diagnoses - hypotonia, cortical vision impairment, global developmental delays, bilateral sensorineural hearing loss, epilepsy, and hypertonic limbs, chronic constipation as well as urology issues. But the journey to find Scarlett’s central diagnosis has been a long one. The basic level of genetic testing routinely performed here in Australia is called a Microarray Analysis. Although Microarray is commonly successful in identifying genetic disorders, and looks for extra (duplicated) or missing (deleted) chromosomal regions, it cannot identify point mutations in a single gene, as is the case for our GNB1 kids. Scarlett had so many tests, ultrasounds, MRIs, a TORCH screen, cerebral spinal fluid analysis, as well as metabolic testing, but it wasn’t until we stumbled across ‘Whole Exome Sequencing’ available in America that we finally found the answer to Scarlett’s challenges. Geneticists here in Western Australia told us we would just have to wait and see …
BUT we couldn’t! We got in touch with project coordinators from 'MyGene2' at the University of Washington, to organize next generation sequencing. After all the tests, some of which invasion, all it took was a saliva sample and we got our answer. There are no words to describe how life changing this was for us! Connecting with other families has been amazing. We are not alone anymore and are working towards targeted therapies and treatment.
The truth is Scarlett has taught us what is important in life. You don’t have to see, you don’t have to hear, walk or talk to be an amazing human being. You only need to love. You only need to feel things right through your soul and smile as if every day is your last. Scarlett is a such a charismatic little girl, she has a heart of gold and is extremely easy going, sociable and affectionate. She is beyond happy and has a smile that lights up our lives every day. We are extremely blessed to have her!
Scarlett is very physically affected by the GNB1 syndrome. She may never be able to roll, sit, crawl, walk, or have perfect head control. But our only hope is that we can give Scarlett every assistance through the GNB1 Foundation so that she can blow us all away with her progress.

Kieren
Master joker, incredible infectious laugh, 7 years, USA
This is our sweet and surprisingly clever 7 year old son Kieren. He is non-verbal and uses an iPad App to communicate. He exceeds his peers in spelling and reading and loves to engage with others, especially if he can get them laughing with his jokes. He uses a manual wheelchair to get around, but loves to climb and go down slides. Kieren is a very hap py and endearing kid who is teaching everyone he meets about how we are all ‘wonders’. We are incredibly proud of him and hope that with the assistance of the GNB1 Foundation, he can have the future he deserves. He might change the world.
Kieren was born in 2011 after a healthy, easy pregnancy. For the first 6 months everything seemed perfect. Of course like most moms do, I remember worrying about his development, but there was a moment I simply knew something wasn’t right. I remember taking him to a baby music class and watching how the little girl next to him moved. She could wriggle and bend and catch herself if she fell from her newfound sitting position. Everyone told me not to worry so much, including our pediatrician who must have thought I was an overly concerned first-time parent. I recall him pulling out textbooks to show me how developmental milestones varied between babies. We were eventually referred to a community early intervention program and a neurologist who both agreed Kieren was delayed, but still weren’t ready to sound the alarm. I was bracing myself for what was to come.
What followed was 6 years of an emotional rollercoaster. During this time we leapt from one possible diagnosis to another. We spent weeks convincing ourselves this precious boy had one devastating disease after another, none of which we had ever heard of until now. We were constantly amazed at how little we knew of the rare diseases and challenges faced by other families. But we were only ever left with ‘no result’ and normal tests, which meant both relief and hopelessness and gathering the energy to continue the search. During this time Kieren endured countless tests, evaluations, referrals, doctor’s appointments and all the while participating in endless therapy.
Finally, when Kieren was 5 years old we started to get some answers. Despite being told for years what we were seeing were not seizures, we pushed for an EEG. This test uncovered a rare form of epilepsy called ESES. They believe this type of epilepsy interrupts slow-wave sleep, which we know is when we rest, recover and consolidate learning. Now for the first time we had a real result and were so hopeful, mainly driven by the hospital’s optimism, that not only had we found the reason for Kieren’s non-verbal, non-mobile developmental path, but there was hope it could be cured. But again after drug trials and tests, nothing changed and we were once again left on our own to fight for our son’s future.
During the 6 years since we had started this journey, genetic testing had come a long way. So with absolutely no belief that we would learn anything, we embarked on tri-xenome sequencing. Meaning they looked at all Kieren’s protein coding genes as well as mine and his dad’s. Shock is a mild term for how I felt the day we got the results … the genetics nurse said Kieren had a missense change in a gene called GNB1 which had recently been implicated in causing many of Kieren’s symptoms: epilepsy, low muscle tone, unusual vision and global developmental delays. While the genetic team couldn’t tell us what this meant for treatment or prognosis, it was such a relief to finally have an answer. I will never be able to fully explain what it means to be able to stop getting up at midnight to research every possible disease you hear about, question whether it is something preventable, wonder whether it is something that I, as his mother, have caused or missed.
Now I can help other families find an answer, now I can do something focused to get my son treatment, quite simply, now I know which of the millions of pages of research and medical papers I might read when I’m up at midnight. Because I’m still up at midnight wondering about the future. The thing that scares me most is the high-risk of devastating seizures and what we still don’t know about how GNB1 will affect Kieren in the long-term. What is wonderful is that the science is now at a point where we can get answers to these questions and our answers might help other families and even the development of better drugs. Thanks to the GNB1 Foundation we have hope.